


A new multimodal generative model co-designs protein sequence and 3D structure from scratch – and produces functional enzymes for reactions with no precedent in natural world.
Enzymes are evolution's master catalysts – molecular machines that accelerate chemical reactions by factors up to a million or more in water, at room temperature. But for all its power, evolution is a tinkerer, not an architect. The enzymes it has produced reflect the chemistry that organisms happened to need – a narrow slice of what proteins could, in principle, catalyze. Vast regions of synthetically valuable chemistry – reactions that could streamline how we manufacture drugs, build materials, and degrade pollutants – remain untouched by biology, not because enzymes cannot perform them, but because no evolutionary pressure ever demanded it.
To date, accessing new enzymatic function has relied on directed evolution – the Nobel Prize-winning strategy of iteratively mutating and screening proteins in laboratories. However, every directed evolution campaign needs a starting enzyme with at least a small amount of the desired activity. For truly new-to-nature reactions, finding that starting point is a challenging process limited by what evolution has already sampled. Meanwhile, deep learning models like RFdiffusion and BindCraft made impressive advances in designing proteins that bind to targets, but designing an enzyme – a protein that does not just hold a molecule but transforms it – is a fundamentally harder problem.
A Single Model for Sequence, Structure, and Function
Today, Caltech, Mila, FutureHouse, and others introduce DISCO (Diffusion for Sequence-structure CO-design); a multimodal generative model that simultaneously designs both the amino acid sequence and the three-dimensional atomic structure of a protein, conditioned on and co-folded with arbitrary biomolecules.
Nearly all experimentally validated protein design pipelines work in two stages: first generate a backbone, then use a separate model to predict a compatible sequence. Because function arises from the inseparable interplay of sequence and structure, this handoff loses information. DISCO instead learns a joint distribution over discrete amino acid tokens and continuous 3D coordinates, denoising both simultaneously. This coupling is enabled by a unified multimodal loss, a cross-modal recycling mechanism, and a self-correcting inference strategy.
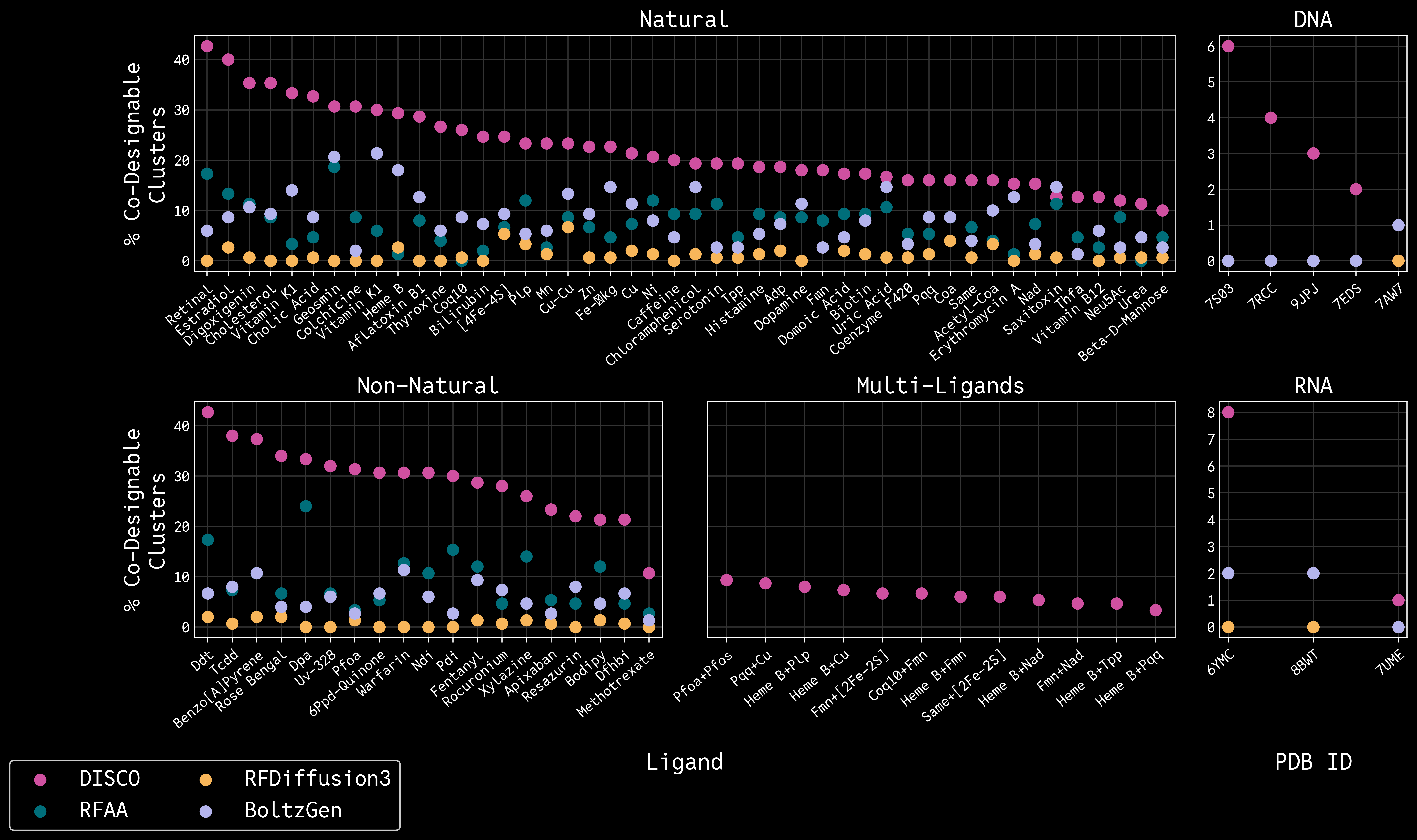
On computational benchmarks, DISCO achieves state-of-the-art performance across diverse design tasks. It generates the most diverse, co-designable protein–ligand complexes for 178 of 179 targets in a new benchmark spanning natural and non-natural molecules, metals, cofactors, DNA, and RNA – outperforming all current baselines.
DISCO further introduces a multimodal inference-scaling method. This allows the generation to explore desirable properties more efficiently than brute-force filtering, including the ability to design (in-silico) for binding specificity between structurally similar molecules.
Designing Enzymes Without a Blueprint
Current enzyme design pipelines require the researcher to specify a "theozyme" before the design begins: the exact geometric arrangement of catalytic residues or the transition state. For new-to-nature chemistry, that mechanistic knowledge is often unavailable. DISCO eliminates this requirement. The model folds a reactive intermediate along with the protein it designs, discovering catalytic solutions on its own.
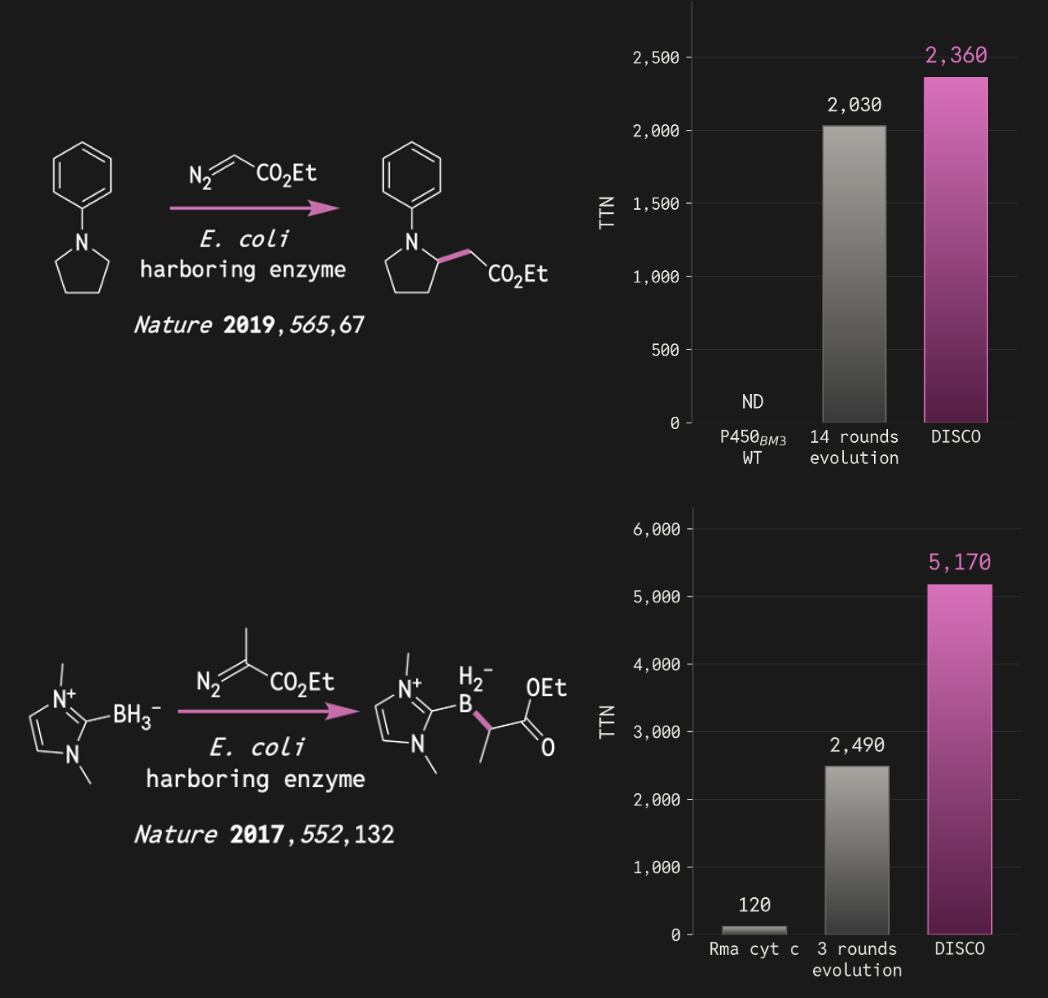
We tested DISCO with carbene-transfer reactions, a class of new-to-nature transformations that have proven valuable for constructing pharmaceutical drugs and complex molecules. We provided no precise theozyme and use no inverse-folding redesign. 90 designs were tested across 4 distinct reactions:
- B–H insertion – a reaction alien to natural world – yielded 98% product and 5,170 total turnovers, more than doubling the activity reached by a previous directed evolution campaign.
- C(sp³)–H insertion - a difficult transformation - reached 2,360 total turnovers, rivaling the outcome of a previous campaign that required 14 rounds of laboratory evolution.
- Alkene cyclopropanation achieved 72% yield and over 4,000 total turnovers with 99:1 diastereoselectivity, surpassing the original evolved enzymes that brought this chemistry into biology.
- Spirocyclopropanation of a pharmaceutically relevant scaffold showed modest initial activity, but a single round of random mutagenesis led to a variety of improved variants with divergent stereoselectivity – suggesting these designs occupy evolvable regions of protein sequence space.
Remarkably, when searched against the AlphaFold Database of over 200 million structures, the active-site geometries of these enzymes have no close natural homologs. None of these designs resemble known heme-binding proteins, either. DISCO did not remix known parts – it invented molecular architectures that evolution never explored, and these architectures are functional. The chemistry nature never explored is now within reach.
For more details, please visit the links below:
Project page: https://disco-design.github.io
Preprint: https://arxiv.org/abs/2604.05181
Code: https://github.com/DISCO-design/DISCO

